
On Wednesday, rapper Albert Johnson—better known by his stage name Prodigy—passed away at the age of 42 from complications of sickle cell disease, an inherited blood disorder he’d had since birth. Though some medications can help those with sickle cell manage their condition, no cure or real treatment exists to combat the disease. And while better care in the United States has extended the lifespan of those with the disease, they often face a lifelong battle with pain, infections, and extreme fatigue.
Sickle cell disease afflicts millions of people worldwide; in the United States alone, about 100,000 live with the blood condition. Someone born with sickle cell disease could only expect to live an average of 14 years in the 1970s, and today the average lifespan still only hits 40 to 50 years.
What causes sickle cell disease?
Sickle cell disease is a term for a group of inherited blood disorders that affect the shape of a person’s hemoglobin—the protein found in red blood cells that carries and delivers oxygen to the rest of the body.
In genetic terms, the disease is autosomal recessive. For a person to develop it, they must receive two different abnormal hemoglobin genes, one from their mother and one from their father. A few different types of these abnormal genes exist, but for it to develop into sickle cell disease, one of these irregular genes must be a type called hemoglobin S. If a person receives two hemoglobin S genes, then they develop what’s called sickle cell anemia, which is the most common and serious disease in the group.
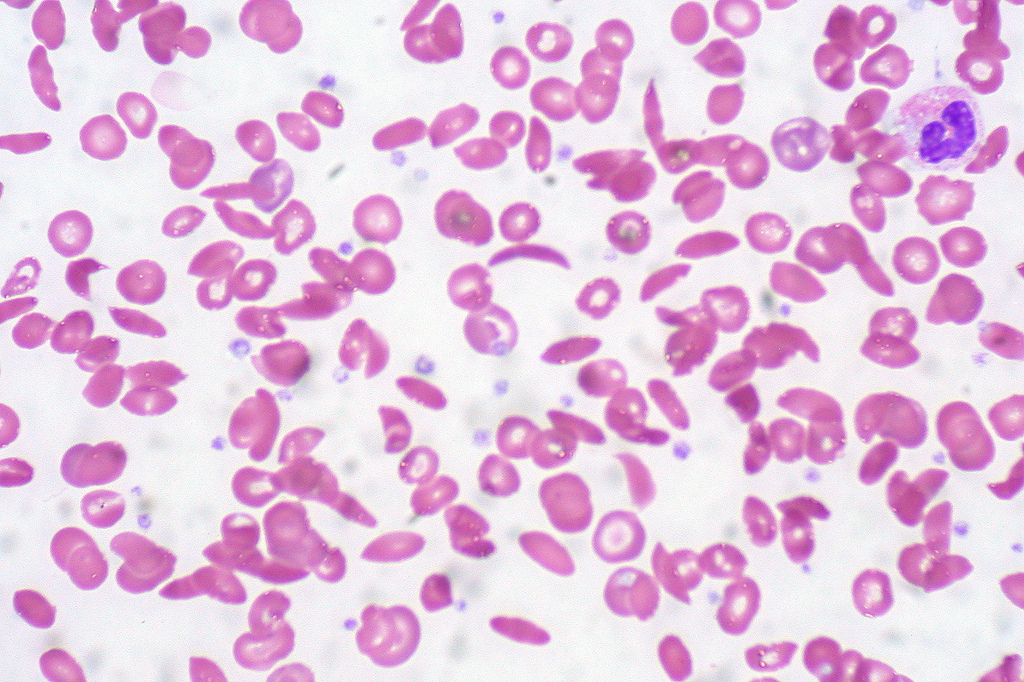
The disease is so debilitating because hemoglobin is one of the most important proteins in the body. When blood reaches the lungs, its job is to collect and transport oxygen to the rest of your organs, which is crucial for survival. Hemoglobin travels inside red blood cells. These cells are typically disc shaped, a form that allows them to easily maneuver en masse through the narrow twists and turns of blood vessels as they travel through the body. But hemoglobin S is weirdly rod shaped, and because of its large size relative to red blood cells, it forces them to take on this oblong shape as well. When every red blood cell is shaped this way, a bunch of them can get jammed inside a blood vessel, slowing or stopping blood flow and preventing oxygen from getting to vital organs. This lack of oxygen can trigger severe pain throughout the body and causes what’s known as a pain crisis, the most common debilitating side effect of the disease. While some people don’t experience much pain, if any, in between these episodes, others live with chronic, ongoing pain throughout their lives. Meanwhile, while a healthy body constantly replenishes its red blood cells, sickle shaped red blood cells tend to die more easily and people with sickle cell disease often aren’t able to keep up with this loss. A lack of red blood cells, known as anemia, can lead to crippling fatigue.
Why is it so much more common in the African-American population?
In the United States, and around the world, the disease is more common in people of African descent. One of the main reasons for this is because people with sickle cell disease, and those with just one copy of the sickle cell trait, have a better chance at surviving malaria, which is common in Africa, than those without the trait. While the exact mechanism of this is not completely clear, researchers do know that because the microorganisms that cause malaria reside in red blood cells, the frequent destruction of sickled red blood cells forces the microorganisms out as well. Sickle cell is a textbook example of an evolutionary phenomenon called balancing selection: genes that can cause sickle cell in pairs are much more likely to persist in populations that get some benefit out of a single copy of the gene—which is only the case in regions plagued by malaria. Within those regions, the benefit of one copy of the gene keeps it from being selected out of the population; the devastating effects of two copies of the gene keep it from becoming too commonplace.
Why haven’t we found a cure? Are we close?
There is currently no cure for the disease. People are often given pain medications and blood transfusions to manage the pain crises and lack of oxygen, but there’s no way to stop the body from making these poorly-shaped hemoglobin cells.
Some people have tried stem cell transplants, where doctors kill off the abnormal hemoglobin with drugs before infusing patients with blood cells from a donor’s bone marrow. Finding a suitable donor is tricky though, and these transplants are especially risky for adults.
However, future treatments may be on the horizon. Because sickle cell disease arises from a mutation in a single gene in a person’s DNA, new gene editing technologies such as CRISPR-Cas9, which allows scientists to edit the human genome with relative ease, could perhaps lead to a way to cure the disease. In fact, this past year, researchers at the University of California, Berkeley, published results from a study showing success in using CRISPR to edit out the disease in mice. However, while promising, we’re still a long way from the treatment reaching humans. The biggest concern is that the editing technique will alter other areas of the genome that appear similar to the ones that need to be edited out—something called off-target effects. Researchers still need to understand why these off-target effects occur, and how to prevent them, before this technique can be safely used in humans.
